User guide¶
So let’s say we have this DNA sequence we want to pretty-print:
>>> sequence = ('cgcactcaaaacaaaggaagaccgtcctcgactgcagaggaagcaggaagctgtc'
... 'ggcccagctctgagcccagctgctggagccccgagcagcggcatggagtccgtgg'
... 'ccctgtacagctttcaggctacagagagcgacgagctggccttcaacaagggaga'
... 'cacactcaagatcctgaacatggaggatgaccagaactggtacaaggccgagctc'
... 'cggggtgtcgagggatttattcccaagaactacatccgcgtcaag')
We can do it with pprint_sequence():
>>> from monoseq import pprint_sequence
>>> print pprint_sequence(sequence)
1 cgcactcaaa acaaaggaag accgtcctcg actgcagagg aagcaggaag ctgtcggccc
61 agctctgagc ccagctgctg gagccccgag cagcggcatg gagtccgtgg ccctgtacag
121 ctttcaggct acagagagcg acgagctggc cttcaacaag ggagacacac tcaagatcct
181 gaacatggag gatgaccaga actggtacaa ggccgagctc cggggtgtcg agggatttat
241 tcccaagaac tacatccgcg tcaag
(This also works if sequence is a Biopython Bio.Seq.Seq object.)
Controlling block and line lengths¶
By default, sequences are printed in blocks of 10 letters, 6 blocks per line. This can be customized with the block_length and blocks_per_line arguments:
>>> print pprint_sequence(sequence, block_length=8, blocks_per_line=7)
1 cgcactca aaacaaag gaagaccg tcctcgac tgcagagg aagcagga agctgtcg
57 gcccagct ctgagccc agctgctg gagccccg agcagcgg catggagt ccgtggcc
113 ctgtacag ctttcagg ctacagag agcgacga gctggcct tcaacaag ggagacac
169 actcaaga tcctgaac atggagga tgaccaga actggtac aaggccga gctccggg
225 gtgtcgag ggatttat tcccaaga actacatc cgcgtcaa g
>>> print pprint_sequence(sequence, block_length=20, blocks_per_line=2)
1 cgcactcaaaacaaaggaag accgtcctcgactgcagagg
41 aagcaggaagctgtcggccc agctctgagcccagctgctg
81 gagccccgagcagcggcatg gagtccgtggccctgtacag
121 ctttcaggctacagagagcg acgagctggccttcaacaag
161 ggagacacactcaagatcct gaacatggaggatgaccaga
201 actggtacaaggccgagctc cggggtgtcgagggatttat
241 tcccaagaactacatccgcg tcaag
Output formats¶
As we’ll see in the next section, certain parts of the sequence can be
annotated for highlighting. For this to work, we need to specify another
output format than the default PlaintextFormat.
monoseq includes three built-in output formats:
PlaintextFormatfor, well, generating plaintext.AnsiFormatadds ANSI escape codes for use in a terminal.HtmlFormatadds HTML tags for inclusion in an HTML document.
(And if this doesn’t satisfy our needs, we can define custom output formats by
implementing Format.)
Formats are specified with the format argument of the
pprint_sequence() function. For example, this is the same example
pretty-printed with HtmlFormat:
>>> from monoseq import HtmlFormat
>>> print pprint_sequence(sequence, blocks_per_line=3, format=HtmlFormat)
<span class="monoseq-margin">1</span> cgcactcaaa acaaaggaag accgtcctcg
<span class="monoseq-margin">31</span> actgcagagg aagcaggaag ctgtcggccc
<span class="monoseq-margin">61</span> agctctgagc ccagctgctg gagccccgag
<span class="monoseq-margin">91</span> cagcggcatg gagtccgtgg ccctgtacag
<span class="monoseq-margin">121</span> ctttcaggct acagagagcg acgagctggc
<span class="monoseq-margin">151</span> cttcaacaag ggagacacac tcaagatcct
<span class="monoseq-margin">181</span> gaacatggag gatgaccaga actggtacaa
<span class="monoseq-margin">211</span> ggccgagctc cggggtgtcg agggatttat
<span class="monoseq-margin">241</span> tcccaagaac tacatccgcg tcaag
As you can see, HtmlFormat wraps the sequence positions in <span>
tags with a class attribute value of monoseq-margin. This allows us to
add custom styling to these numbers with a CSS stylesheet.
Note
In an HTML document, include the pretty-printed sequence within
<pre> and </pre>. This preserves all whitespace and automatically
selects a monospace font.
Sequence annotations¶
Subsequences can be highlighted in the pretty-printed sequence by specifying their positions. Such a specification is called an annotation. Several annotations can be provided and each of them will be highlighted in a distinct style (e.g., the first annotation is colored red and the second is printed in bold).
Let’s assume our analysis shows positions 12 through 37 and 223 through 247 to be highly conserved between species. Of course, we want to annotate our sequence with this knowledge:
>>> from monoseq import AnsiFormat
>>> conserved = [(11, 37), (222, 247)]
>>> print pprint_sequence(sequence, format=AnsiFormat,
... annotations=[conserved])
1 cgcactcaaa acaaaggaag accgtcctcg actgcagagg aagcaggaag ctgtcggccc
61 agctctgagc ccagctgctg gagccccgag cagcggcatg gagtccgtgg ccctgtacag
121 ctttcaggct acagagagcg acgagctggc cttcaacaag ggagacacac tcaagatcct
181 gaacatggag gatgaccaga actggtacaa ggccgagctc cggggtgtcg agggatttat
241 tcccaagaac tacatccgcg tcaag
Note
Regions are defined as in slicing notation, so zero-based and open-ended.
Just for lack of imagination, we also want to make it clear where every 12th
nucleotide is in our sequence. We can do this by defining a second
annotation, which is printed in bold by AnsiFormat:
>>> twelves = [(p, p + 1) for p in range(11, len(sequence), 12)]
>>> print pprint_sequence(sequence, format=AnsiFormat,
... annotations=[conserved, twelves])
1 cgcactcaaa acaaaggaag accgtcctcg actgcagagg aagcaggaag ctgtcggccc
61 agctctgagc ccagctgctg gagccccgag cagcggcatg gagtccgtgg ccctgtacag
121 ctttcaggct acagagagcg acgagctggc cttcaacaag ggagacacac tcaagatcct
181 gaacatggag gatgaccaga actggtacaa ggccgagctc cggggtgtcg agggatttat
241 tcccaagaac tacatccgcg tcaag
AnsiFormat supports up to three annotation levels and the third one
is printed underlined. So if the middle third of the sequence would be our
primary concern, we could underline it as follows:
>>> middle = [(len(sequence) / 3, len(sequence) / 3 * 2)]
>>> print pprint_sequence(sequence, format=AnsiFormat,
... annotations=[conserved, twelves, middle])
1 cgcactcaaa acaaaggaag accgtcctcg actgcagagg aagcaggaag ctgtcggccc
61 agctctgagc ccagctgctg gagccccgag cagcggcatg gagtccgtgg ccctgtacag
121 ctttcaggct acagagagcg acgagctggc cttcaacaag ggagacacac tcaagatcct
181 gaacatggag gatgaccaga actggtacaa ggccgagctc cggggtgtcg agggatttat
241 tcccaagaac tacatccgcg tcaag
Styling HtmlFormat output¶
The HtmlFormat output format supports up to 10 annotation levels, but
how to style them is up to the user. All monoseq does is add <span>
tags around annotations with class attribute values of
monoseq-annotation-{i}, where {i} is the annotation level starting
from 0.
Here are some example CSS rules for styling 4 annotation levels:
pre {
background: lightYellow;
color: black;
}
.monoseq-margin {
color: grey;
}
.monoseq-annotation-0 {
color: red;
}
.monoseq-annotation-1 {
background: black;
color: lightYellow;
}
.monoseq-annotation-1 .monoseq-annotation-0 {
background: red;
color: lightYellow;
}
.monoseq-annotation-2 {
text-decoration: underline;
}
.monoseq-annotation-3 {
font-weight: bold;
}
Using these rules, a pretty-printed protein sequence will look something like this:
1 MIMANQPLWL DSEVEMNHYQ QSHIKSKSPY FPEDKHICWI KIFKAFGMIM ANQPLWLDSE 61 VEMNHYQQSH IKSKSPYFPE DKHICWIKIF KAFGMIMANQ PLWLDSEVEM NHYQQSHIKS 121 KSPYFPEDKH ICWIKIFKAF GMIMANQPLW LDSEVEMNHY QQSHIKSKSP YFPEDKHICW 181 IKIFKAFG
Using monoseq from the IPython Notebook¶
For pretty-printing sequences directly in the IPython Notebook, monoseq.ipynb.Seq is provided as a
convenience wrapper around pprint_sequence():
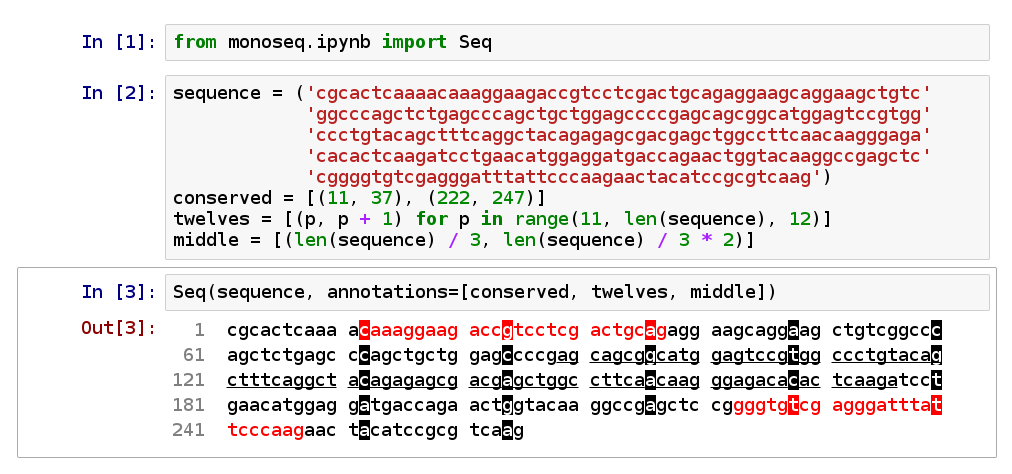
This supports up to four levels of annotation, displayed as red, inverted, underlined, and bold.
See this IPython Notebook for some examples.